Lots of variation exists on this topic, realistically because of the variations in the properties of amino acids (this is stating the obvious). Not so obvious is the impact of the color schemes given to the amino acids, on the individual 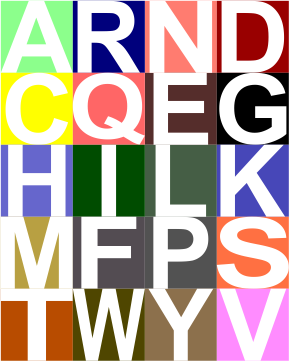 researchers who observe them, as they try to figure out if they have meaning. They respond to subconscious “emotions” and “thoughts” totally unrelated to science, and the colors are conveying “information” about which individuals are unaware.
researchers who observe them, as they try to figure out if they have meaning. They respond to subconscious “emotions” and “thoughts” totally unrelated to science, and the colors are conveying “information” about which individuals are unaware.
Case in point, as i looked over the color scheme for amino acids from RCSB, I saw no real order or purpose. Some colors don’t even look nice, and in one case there was a duplicate (since corrected). Also since these colors are no longer what RCSB uses, here comes a new diagram.
A venn diagram described by its maker thusly: “Venn diagram grouping of amino acids based on the biochemical properties: hydrophobicity, size and polarity. Colors for the groups are derived by additive blending of the colors of representative properties.” The blending of colors to create intermediate colors could be better in my “visual” opinion, and I will have to research just how the RGB, or CMYK, or HSL, or whatever they used was calculated (original image uploaded by uploaded by Florian Battke to Research gate). I assume from the venn diagram that the following is more or less what is proposed:
Oranges = hydrophobic
Blue = small
Greens = polar
Blue-green = negative
Turquoise = small polar
Grey = small negative hydrophobic
Pink = small hydrophobic
Brown-orange = hydrophobic aromatic
Orange-pink = hydrophobic aliphatic
This swatch with letter ID is alphabetical, unlike their original which did go by color.

This approach (ref http://www.biomedcentral.com/1471-2105/13/S8/S) calls the method (integrated hierarchical aggregation tables for sequence alignment (with an unfortunate similar acronym with some group about iraq) sorts and displays