1 (ONE) molecule (AFM image of surfactant protein D) which I call 41_aka_45 ( published by Arroyo et al, 2018)

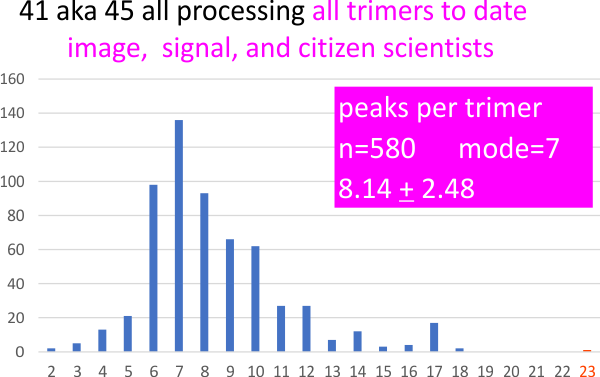
SUMMARY: 159 different grayscale (LUT) plots of one surfactant protein D dodecamer — as 2 hexamers (arm 1, arm 2) and as 4 trimers (arm 1a, 1b and arm 2a and 2b) show that personal judgement is still critical for determining the number of brightness peaks along this molecule.
METHODS: 12 image processing programs (listed below) were used to filter, mask, limit range, change contrast, HSL, etc, to enhance the appearance of peaks in this image.
2 programs (listed below) were used for plotting grayscale data (ImageJ (used on almost all images) and Octave/Matlab (occasionally).
5 signal processing programs were used to count the number of peaks in grayscale plots made by a 1px segmented line using imageJ, with dozens of variations in the input statements for those signal processing programs.
12 peak counts were obtained from volunteers for a single set of plots of this dodecamer (ages 8 – 74 volunteer citizen scientist impressions of the number of peaks).
DATA: All data were saved in an excel file with all image and signal processing parameters to allow assessment of combinations of processing programs and types to produce the most convincing peak counts, widths, and heights.
PURPOSE: To identify a method(s) for assessing the number of peaks, relative peak widths and heights for AFM images of surfactant protein D. (A method that could be applied to countless other AFM application and other molecules).
RESULTS: Nothing produced the results I had hoped for, but there is a clear trend in peak number, width and height. (See next post sometime in the future).
CONCLUSION: Variations in the number of peaks detected in each hexamer produce both an even number or odd number of peaks, the mean and mode are similar, for hexamers (mean=15 peaks, median=15 peaks, mode is something closer to 13 peaks). The likely peak number for any given surfactant protein D hexamer is still open, since this is an analysis of methods, not molecules. The use of this molecule was based on observations of about 100 other images, and represents a reasonable “good choice” to select a methodology. There are clear options for the most efficient peak detection in image and signal processing, and there are just as clear deficiencies.
A summary of the number of programs, plots and peaks applied to this one molecule is shown below – and it represents the sum total of the data image processing, signal processing, and quick peak counts from citizen scientists, as well as the 100 or so counts of my own, from each image.
Abbreviations:
Image processing programs: psd=Photoshop (proprietary, Photoshop 6 and Photoshop 2021; cpp=corelPhotoPaint (proprietary – raster graphics program, CorelPhotoPaint x5 and 2019); cdr=corelDRAW (proprietary vector graphics program, CorelDRAW x5 and 2019 where the image adjustment menu was used); gw=Gwyddion, a multiplatform modular free software for visualization and analysis of data from scanning probe microscopy techniques, used here ONLY for image processing; paint=Paint.net (free, open source raster graphics program); gimp=GIMP GNU Image Manipulation Program (free, opensource); inkscape= Inkscape.org (free and open source) vector graphis program; Octave/Matlab (Octave is free and open source) used briefly for image processing (separate from signal processing, limited to 3 plots total, as this was a super cumbersome way to process images). ImageJ, used for both image processing and excel plots (ImageJ is a Java-based image processing program developed at the National Institutes of Health and the Laboratory for Optical and Computational Instrumentation)(free). I added a column of counts of my own, made from each processed image as well (my peak counts).
Signal processing apps: batchprocess (Aaron Miller’s app for batch processing excel files using the Lag, Threshold, Influence (open source library); Octave/Matlab (various settings for FindPeaksPlot, AutoFindPeaksPlot, Ipeaks)(check out Thomas O’Haver’s website); scipy, (Daniel Miller’s app for peak finding using Prominence Distance Width Threshold Height (Sci/Python open source library); Two excel peak finding templates – 1) PeakDetectionTemplate.xlsx and 2) PeakValleyDetectionTemplate.xlsx (Thomas O’haver). Many variations for amplitude threshold, slope threshold, lag, distance, width, smoothing and many others) were used in signal processing.
Citizen science: Peak counts in a single set of plots of this dodecamer were obtained from a group of friends and family, ages 8 – 78. I did not include my counts in this category as they numbered in the hundreds, not just one set of plots as the former.
Below is just a summary of the number of trimers plotted, in each of the above image and signal processing programs.

Summary of all counts of 2 hexamers, one dodecamer

My counts from the image as processed dozens of times with dozens of filters with the 12 vector and raster imaging programs produced the most consistent results, but very similar to my own counts of the actual excel plots generated by a trace through the center of the hexamers (2 trimers) were found in a manual count of the peaks of plots made in ImageJ. Both image processing filters and signal processing algorithms have a huge impact on the variation (var) and the min, and max of the number of peaks counted (judgement is required).
It is worth noting that my counts of peaks from the images is the the lowest, meaning to me that processing might be a good backup for confirming what is seen by eye. – In fact, the reason for this study was that I saw a pattern in the peaks (mine more detailed and specific than the pattern reported in the literature), and I wondered if it was provable.
I doubt adding more peak counts to this data is going to change much (LOL). So now, the approach is to separate out the filters, masks, and algorithms which best fit the mean and median.