8 dodecamersL SP-D: Subpeaks per peaks detected using AFM images of surfactant protein D. (APOLOGETICS) Always and at the outset I thank Arroyo et al for the 2020, 2018 publication of the SP-D AFM images (the best I have encountered of SP-D), and secondly I thank Dan Miller for the scipy app for peak finding, Aaron Miller for the LTI app for peak finding and batch processing, and Thomas O’Haver for his help with Octave, and excel templates, and also for ImageJ and Gwyddion (and I guess i should be very grateful for the original developers of CorelDRAW (which was Kodak)(not the new owners as they get a thumbs down from me), and also the original producers of Photoshop (yep, version 6 on CDs performed just as well for image analysis as rented versions of 2021 (so it is also thumbs down to them)).
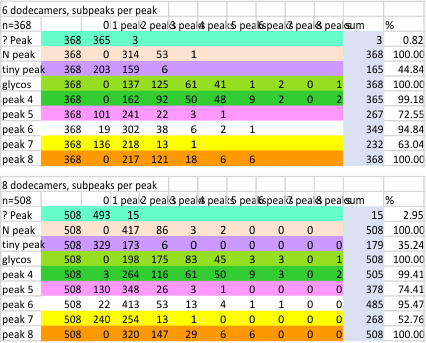
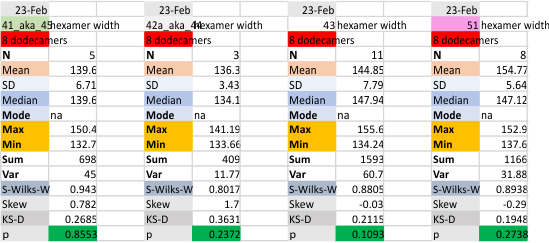
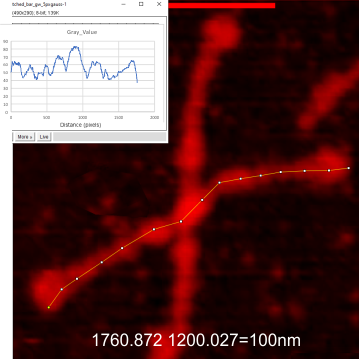
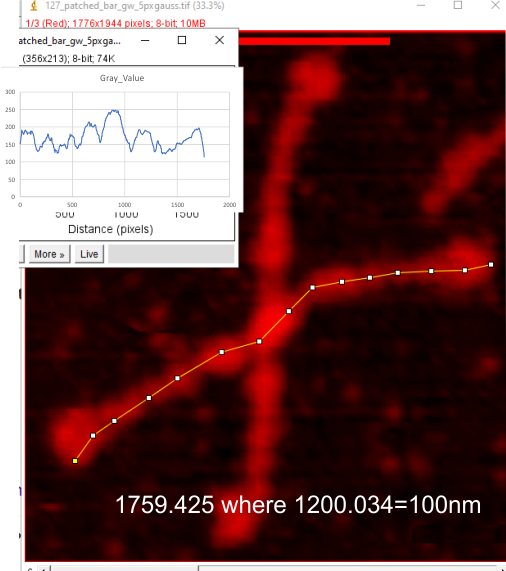
The following is the result of 508 image and signal processing plots of 8 images of surfactant protein D. These data are reported as individual plots of hexamers (thus four trimer plots (as separate entities, with plots beginning at the full width of the N term plot and progressing to the CRD. I have made the assumption (which I will discuss) that signal processing algorithms are smart enough to see symmetry… bilateral symmetry, which apparently is giving that AI too much credit.
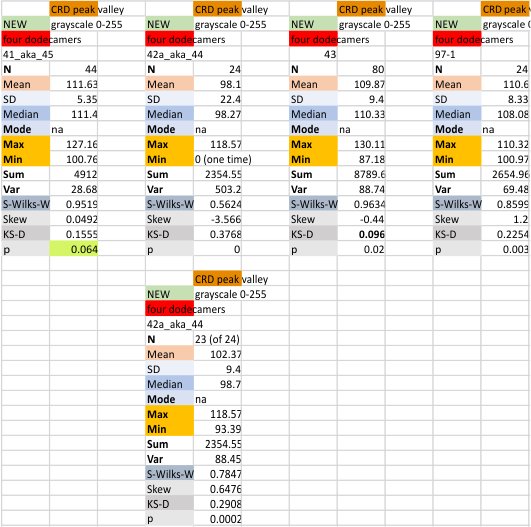
Notwithstanding that problem, the total number of peaks per trimer (8) established some time ago is the number which is used to box the number of subpeaks into 8. Below are the data for 6 dodecamers (n = number of plots analyzed, not the number of dodecamers analyzed) and 8 dodecamers. Consistency is apparent. Not all peaks show up 100% of the time. Peaks such as the N and glycosylation and peak 5 and CRD peaks are often lumpy (meaning they have subpeaks.
There is a peak called “?” which only rarely occurs in dodecamers (but in my opinion is frequent in multimers (called fuzzy balls) and is indicative of a side to side N term association among molecules. It is reasonable for that peak NOT to show up below.
The N peak is present 100% of the time, as is the CRD peak and the glycosylation peak (though the height of the glycos peak varies (and at this point unglycosylated AFM images of SP-D have not been analyzed, so that will be dependent on the SP-D molecule, which species, and mutations and other factors, but here it is rhSP-D). Peak 4 is very consistent, present 99+ percent of the time, not previously reported, lying in the collagen-like domain. The next two peaks have characteristics that are obvious visually, peak 5 is not wide, and is not tall but consistently shows up right after peak 5. Peak 6 is broad, and appears regularly (94+ percent of the time, and is also low. Peak 7 is what I believe is the neck of the SP-D trimer, and it is very often covered by the bright peak of the CRD (and this depends on whether the rounded ball shaped CRD peaks are positioned directly over the neck or to one side. (Just my opinion here). The glycosylation peak and peak typically have more than one subpeak.