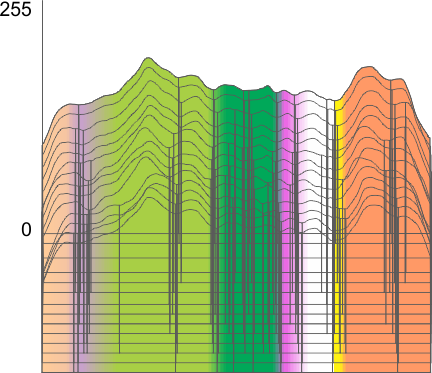
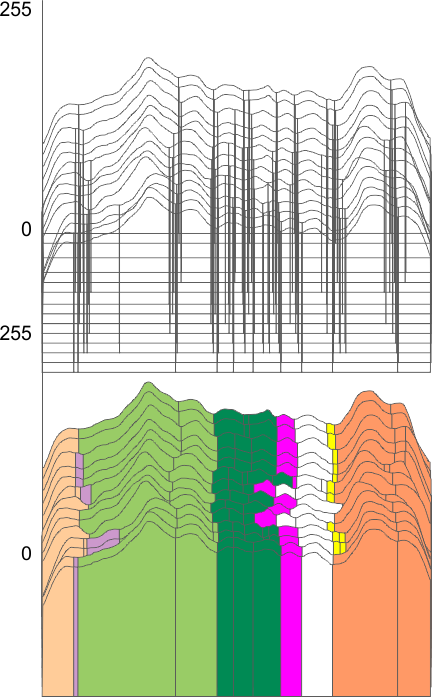
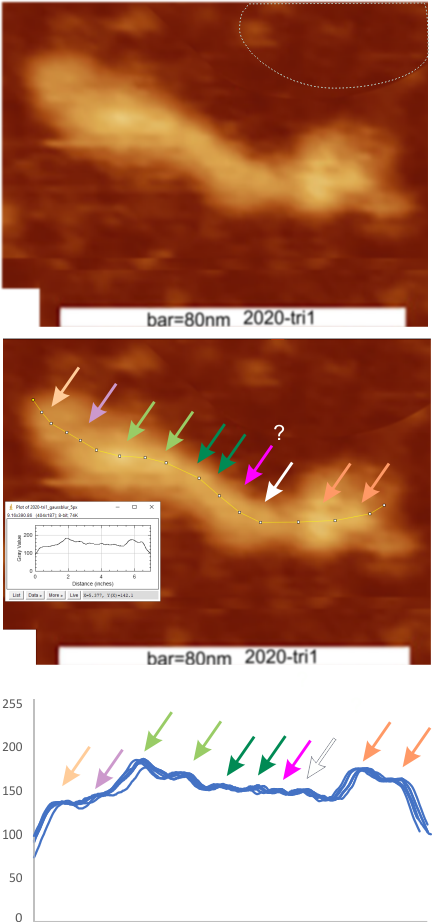
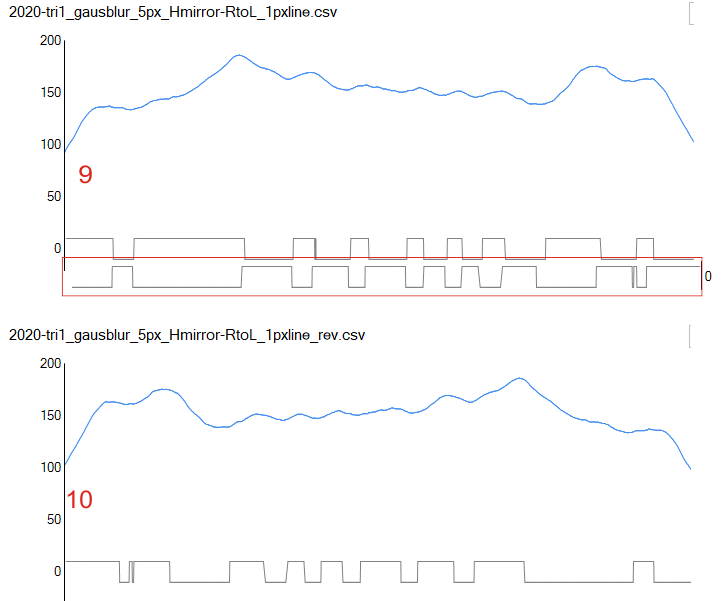
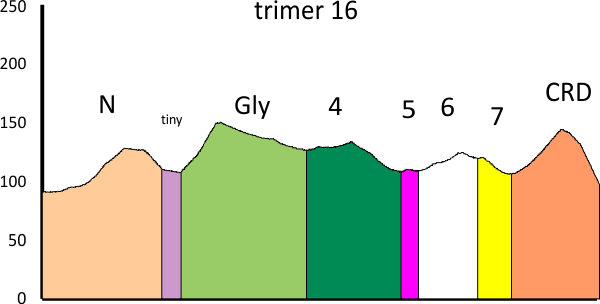
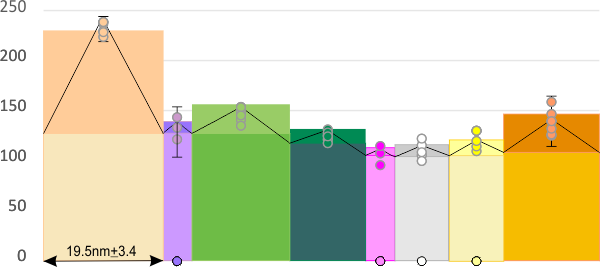
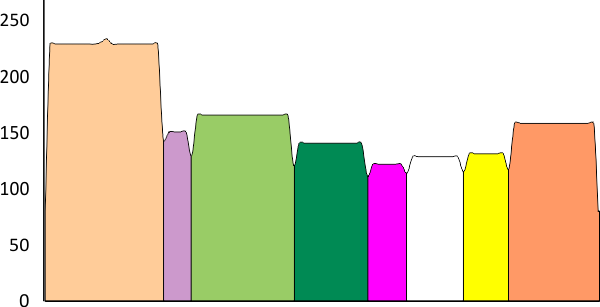
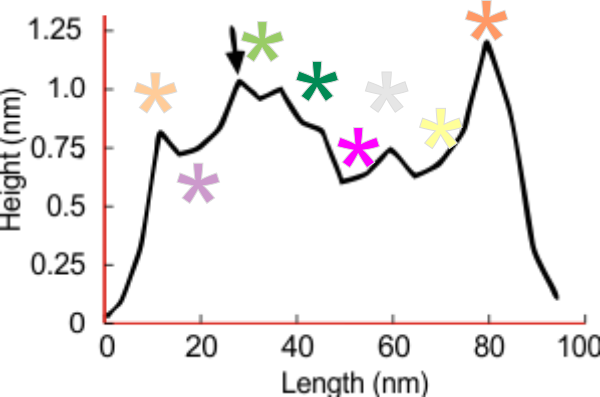
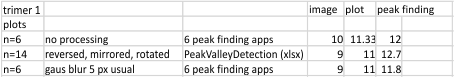Using the PeakValleyDetectionTemplate.xlsx from Tom O’Haver, I ploted a single SP-D trimer, forward blackward, mirrorred, reversed using excel, as well as bottom to top and reversed. (PeakValleyDetectionTemplate.xlsx, smooth 11. Peak identification on SP-D trimer shows very little change in peak number, width, height and valley (as determined by grayscale plots made using ImageJ) no matter how I enter the csv file. This is good in a way, as it means the high peaks next to the low peaks, at (smooth 11) pretty much are read the same way, front to back and back to front.
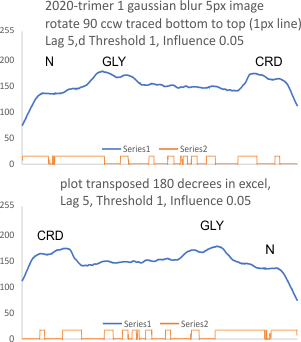
The greater variation comes from image processing and variation in my trace through the center width of the molecule from N term peak to CRD peak(s) at least that seems to be true with this template at smooth 11.
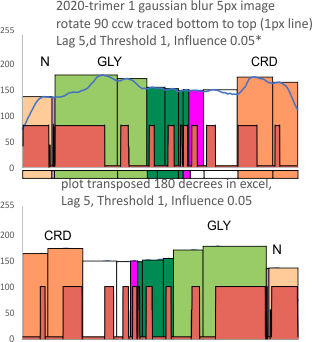
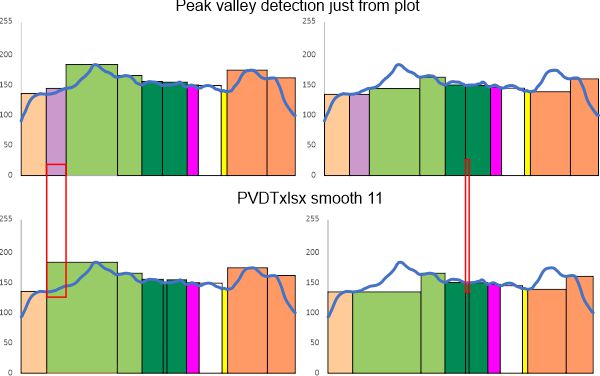
In the graphic below columns on the right hand side are “image” (= my peak count), “plot” (=my peak count from the ImageJ grayscale plot). “peak finding” (= peaks found using signal processing apps – mentioned many times in previous posts). the center row of data for peak numbers is derived JUST from the PVDTxlsx peak numbers.
Rows 3 and 6 are plots of the same trimer using the unprocessed image and gaussian blur (5px) image respectively.
I do think the number of peaks per trimer will turn out to be “8” as these numbers are an n of 1, just to determine whether the peak finding xlsx plots are influenced by direction of the plot line… The image used for these plots was particularly nice and that would be selection bias toward more peaks.