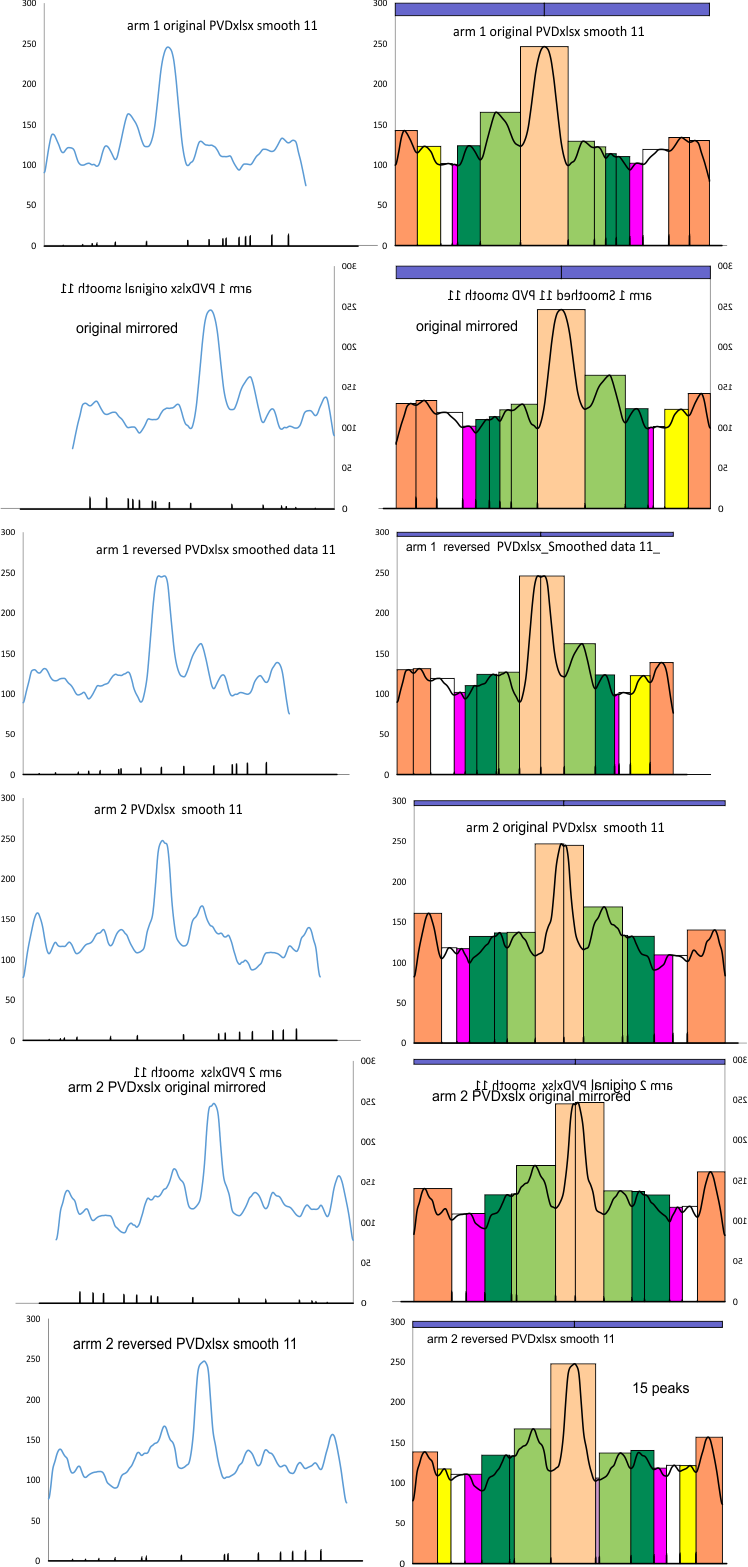
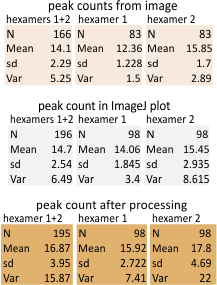
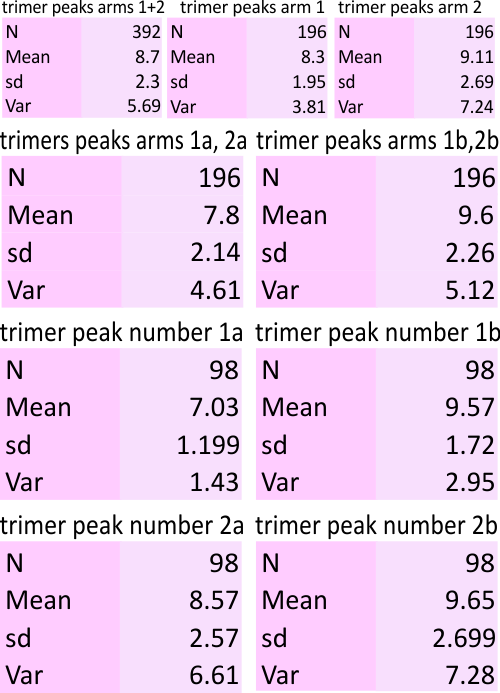
11 peaks (brightness/grayscale) traced along a center, 1px line in a surfactant protein D hexamer is the absolute minimum that can be easily described. Furthermore, and routinely, as many as 15 peaks can be found. More are seen with different algorithms, but may not be consistent. Much depends upon how the molecule falls, the over or undersaturation of the contrast (in the original and in the image processing), and the quality of the image (obviously). Also, critical to finding peaks is the care with which the segmented line is drawn (the greater attention to this is what produces two peaks (sometimes 3) in the center of the N termini junction, and in the area just before the N termini junction (i.e. the area between the glycosylation(s) site and the N termini – which i have referred to in several posts as the “tiny peak”).
Thanks to Arroyo et. al (2018). (about 90 images of SP-D) and Thomas 0’Haver and Aaron Miller and Daniel Miller (numerous signal processing programs and algorithms) for their contributions of numerous signal processing programs and algorithms.
11 peaks is just the simplest count from poorer resolution AFM images, using just the most basic image and signal processing tools which are freely available to anyone. But careful analysis and fine tuning images with gaussian, median, mean, sharpening, and range limiting filters, as well as optimizing options such as smoothing, lag, threshold, width, influence, etc in signal processing shows the peak number to be close to 15+ per hexamer.
One of the surprises of signal processing plots from ImageJ is an apparent disadvantage in peak detection in being a molecule with large differences in peak brightness, coupled with prominent bilateral symmetry.
In the sense of perception of mirror symmetry has significant functions human perception. It has been shown that symmetry is detected within randomly placed elements, and it would be an easy leap to see this visual function as evolutionarily advantageous. Whats more, subjectively speaking, symmetry is apparently pleasing (using self as a sample) as I do this in my own artwork, and see it in architecture and other artwork as well. Not only symmetry but repetition (particularly iterations with rotation) and nature does this supremely well, and it has been studied mathmatically. Instances are too numerous to list and readily available to search online.
An interesting attribute of human sensitivity to mirror symmetry is “tolerance” to error, meaning that variations dont matter a whole lot. This may relate to the extreme abuncance of what biology does with duplicate molecules in arranging them into dimers, trimers, etc on ad nauseum. I am thinking that my interest in surfactant protein D, which has so much “symmetry” (not unlike other patterned, replicated, duplicated, inverted, rotated, molecules found everywhere), and just enough noise may be a visual symmetry puzzle, at best.
What is exciting is that SP-D runs the gamut of everything from monomer to multimer (multimer here being that unique molecule named the “fuzzy ball” by some surfactant protein D researchers, which at any point can have 100 or more monomers). In time, such a multimer will likely show numerous mirror, rotate, iterations which can be seen at quick glance before any image or signal processing. Just for fun I re-uploaded a graphic mandala that I made a couple years ago, which is a “fake” look at fuzzy ball symmetry (12 trimers, 36 monomers. Alleged glycosylation peaks are a dark blue ring arou the center, CRD are the bumps at the ends of the molecules. Actual SP-D image, masked and colored in CorelDRAW. Pink background, black, white and blue abstract borders are “fills” and not relevant to SP-D structure. Artistically speaking i should go back and edit the background and border for more pleasing texture tile settings and colors. (LOL) and rearrange in the trimers. Even in this artsie craftsie image the peaks along the arms can be seen. I am not ready yet to relegate the entire interpretation of the shape of SP-D to the best image and signal processing programs. Human input is obviously required still.